









Inhoud
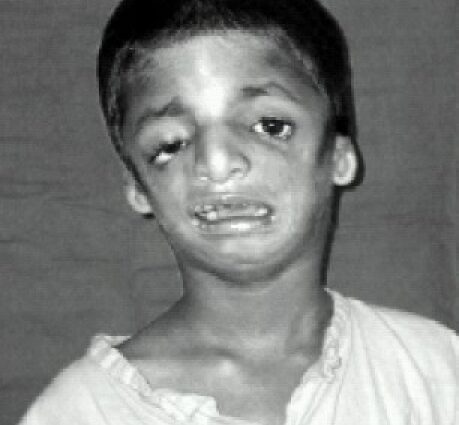
Treacher-Collins-syndroom
Een zeldzame genetische ziekte, het Teacher-Collins-syndroom, wordt gekenmerkt door de ontwikkeling van geboorteafwijkingen van de schedel en het gezicht tijdens het embryonale leven, resulterend in misvormingen van het gezicht, de oren en de ogen. De esthetische en functionele gevolgen zijn min of meer ernstig en in sommige gevallen zijn talrijke chirurgische ingrepen nodig. Maar in de meeste gevallen zorgt het nemen van regie ervoor dat een bepaalde kwaliteit van leven behouden blijft.
Wat is het Treacher-Collins-syndroom?
Definitie
Treacher-Collins-syndroom (genoemd naar Edward Treacher Collins, die het voor het eerst beschreef in 1900) is een zeldzame aangeboren ziekte die zich vanaf de geboorte manifesteert met min of meer ernstige misvormingen van het onderste deel van het lichaam. gezicht, ogen en oren. De aanvallen zijn bilateraal en symmetrisch.
Dit syndroom wordt ook wel Franceschetti-Klein syndroom of mandibulo-faciale dysostose zonder eindafwijkingen genoemd.
Oorzaken
Tot nu toe is bekend dat drie genen betrokken zijn bij dit syndroom:
- het TCOF1-gen, gelokaliseerd op chromosoom 5,
- de POLR1C- en POLR1D-genen, die zich respectievelijk op chromosomen 6 en 13 bevinden.
Deze genen sturen de productie van eiwitten aan die een sleutelrol spelen in de embryonale ontwikkeling van gezichtsstructuren. Hun verandering door mutaties verstoort de ontwikkeling van botstructuren (voornamelijk die van de onder- en bovenkaak en jukbeenderen) en zachte weefsels (spieren en huid) van het onderste deel van het gezicht tijdens de tweede maand van de zwangerschap. Ook de oorschelp, de gehoorgang en de structuren van het middenoor (gehoorbeentjes en/of trommelvliezen) worden aangetast.
Diagnostisch
Aangezichtsmisvormingen kunnen worden vermoed vanaf de echografie van het tweede trimester van de zwangerschap, vooral in gevallen van significante oormisvormingen. In dit geval zal de prenatale diagnose worden gesteld door een multidisciplinair team van magnetische resonantie beeldvorming (MRI) van de foetus, waardoor de misvormingen nauwkeuriger kunnen worden gevisualiseerd.
Meestal wordt de diagnose gesteld door een lichamelijk onderzoek bij de geboorte of kort daarna. Vanwege de grote variabiliteit van misvormingen, moet het worden bevestigd in een gespecialiseerd centrum. Een genetische test op een bloedmonster kan worden besteld om te zoeken naar de betrokken genetische afwijkingen.
Sommige milde vormen blijven onopgemerkt of worden toevallig laat ontdekt, bijvoorbeeld na het verschijnen van een nieuw geval in de familie.
Zodra de diagnose is gesteld, wordt het kind onderworpen aan een reeks aanvullende onderzoeken:
- gezichtsbeeldvorming (röntgenfoto, CT-scan en MRI),
- ooronderzoeken en gehoortesten,
- visie beoordeling,
- zoeken naar slaapapneu (polysomnografie) …
De betrokken mensen
Het Treacher-Collins-syndroom treft naar schatting één op de 50 pasgeborenen, zowel meisjes als jongens. Naar schatting komen er in Frankrijk elk jaar ongeveer 000 nieuwe gevallen bij.
Risicofactoren
Erfelijkheidsadvies in een verwijscentrum wordt aanbevolen om de risico's van genetische overdracht te beoordelen.
Ongeveer 60% van de gevallen komt geïsoleerd voor: het kind is de eerste patiënt in het gezin. Misvormingen treden op na een genetisch ongeval waarbij een van de voortplantingscellen die bij de bevruchting betrokken zijn, is aangetast (“de novo”-mutatie). Het gemuteerde gen wordt dan doorgegeven aan zijn nakomelingen, maar er is geen bijzonder risico voor zijn broers en zussen. Er moet echter worden gecontroleerd of een van zijn ouders niet daadwerkelijk lijdt aan een lichte vorm van het syndroom en de mutatie draagt zonder het te weten.
In andere gevallen is de ziekte erfelijk. Meestal is het risico van overdracht één op twee bij elke zwangerschap, maar afhankelijk van de betrokken mutaties zijn er andere wijzen van overdracht.
Symptomen van het Treacher-Collins-syndroom
De gelaatstrekken van de getroffenen zijn vaak karakteristiek, met een geatrofieerde en terugwijkende kin, niet-bestaande jukbeenderen, ogen die naar beneden gericht zijn naar de slapen, oren met een klein en slecht omzoomd paviljoen, of zelfs volledig afwezig …
De belangrijkste symptomen zijn gekoppeld aan misvormingen van de KNO-bol:
Ademhalingsproblemen
Veel kinderen worden geboren met smalle bovenste luchtwegen en nauw geopende monden, met een kleine mondholte die grotendeels wordt belemmerd door de tong. Vandaar aanzienlijke ademhalingsmoeilijkheden, vooral bij pasgeborenen en zuigelingen, die tot uiting komen in snurken, slaapapneu en een te zwakke ademhaling.
Moeilijkheden met eten
Bij zuigelingen kan borstvoeding worden belemmerd door ademhalingsmoeilijkheden en door afwijkingen van het gehemelte en het zachte gehemelte, soms gespleten. Eten is gemakkelijker na de introductie van vast voedsel, maar kauwen kan moeilijk zijn en gebitsproblemen komen vaak voor.
Doofheid
Gehoorproblemen als gevolg van misvormingen van het buiten- of middenoor zijn in 30 tot 50% van de gevallen aanwezig.
Visuele stoornissen
Een derde van de kinderen lijdt aan scheelzien. Sommige kunnen ook bijziend, hyperopisch of astigmatisch zijn.
Leer- en communicatieproblemen
Treacher-Collins-syndroom veroorzaakt geen intellectuele achterstand, maar doofheid, visuele problemen, spraakmoeilijkheden, de psychologische gevolgen van de ziekte en de stoornissen veroorzaakt door vaak zeer zware medische zorg kunnen vertraging veroorzaken. taal en moeite met communiceren.
Behandelingen voor het Treacher-Collins-syndroom
Kindzorg
Ademhalingsondersteuning en/of sondevoeding kunnen nodig zijn om de ademhaling en voeding van de baby te vergemakkelijken, soms vanaf de geboorte. Wanneer ademhalingshulp in de loop van de tijd moet worden gehandhaafd, wordt een tracheotomie (kleine opening in de luchtpijp, in de nek) uitgevoerd om een canule in te brengen die direct zorgt voor de doorgang van lucht in de luchtwegen.
Chirurgische behandeling van misvormingen
Min of meer complexe en talrijke chirurgische ingrepen met betrekking tot het zachte gehemelte, kaken, kin, oren, oogleden en neus kunnen worden voorgesteld om het eten, ademen of horen te vergemakkelijken, maar ook om de esthetische impact van misvormingen te verminderen.
Ter indicatie: de spleten van het zachte gehemelte worden gesloten vóór de leeftijd van 6 maanden, de eerste cosmetische ingrepen aan de oogleden en jukbeenderen vanaf 2 jaar, de verlenging van de onderkaak (mandibulaire distractie) naar 6 of 7 jaar, herbevestiging van de oorschelp rond de 8 jaar, vergroting van de gehoorgang en/of operatie van de gehoorbeentjes rond de 10 tot 12 jaar… Andere cosmetische chirurgische ingrepen kunnen nog uitgevoerd worden in de adolescentie…
Hoorapparaat
Gehoorapparaten zijn soms mogelijk vanaf de leeftijd van 3 of 4 maanden wanneer doofheid beide oren aantast. Er zijn verschillende soorten prothesen beschikbaar, afhankelijk van de aard van de schade, met een goede efficiëntie.
Medische en paramedische opvolging
Om arbeidsongeschiktheid te beperken en te voorkomen is de reguliere monitoring multidisciplinair en doet een beroep op verschillende specialisten:
- KNO (hoog risico op infectie)
- Oogarts (correctie van gezichtsstoornissen) en orthoptist (oogrevalidatie)
- Tandarts en orthodontist
- Spraaktherapeut…
Vaak is psychologische en educatieve ondersteuning nodig.